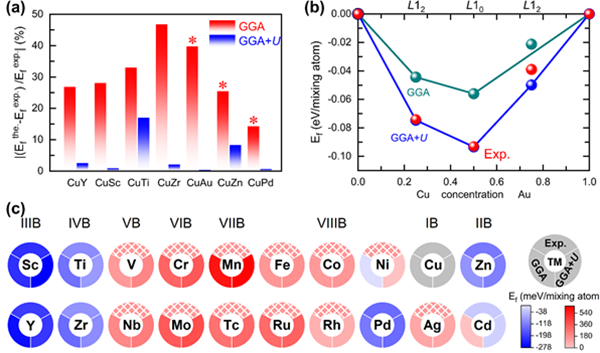
铜基过渡金属(Cu-TMs)是实验研究最多的金属间化合物之一,在催化、电子元件和基础冶金等领域有重要的应用价值。其中,Cu-Au体系是研究合金的相图、电子结构、有序-无序转变等性质的典型范例。然而,使用基于广义梯度近似的密度泛函理论(DFT-GGA)却不能准确模拟Cu-Au合金的性质:(1) GGA预测的形成能与实验测量值之间存在较大误差;(2) GGA错误地预测了富Au基态。该工作中,研究人员发现这些差异也广泛存在于其它Cu-TMs合金当中,这严重阻碍了DFT-GGA在合金体系中的应用。因此,阐明DFT-GGA错误预测Cu-TMs化合物热力学性质的内在机制并寻找正确模拟合金形成能等物性的方法具有重要意义。
为此,研究人员探究了7种有序合金的形成能,发现由Cu和含d 电子数较少的过渡金属(Sc、Y、Ti、Zr)组成的合金,其形成能在GGA计算中会被高估;而由Cu和含d 电子数较多的金属(Au、Cu、Pd)组成的合金,其形成能则被低估,这表明理论计算的形成能对d 带能级分布极其敏感。进一步研究表明,在过渡金属(TM)单质中,GGA可以准确模拟出在费米能级处有d 电子贡献的TM-d 带的能量分布范围,而不能正确给出d 带分布较深的TM-d 带的能量分布范围。通过采用广义梯度近似结合Hubbard U 修正(GGA+U )方法,将金属的d 带能级调控至实验报道的能量区间(-6 eV 到 -2 eV),准确地模拟了合金中Cu与Au原子间d 带的相互作用,从而能够准确地获得形成能与富Au基态结构。此外,研究人员还深入研究了另外19种Cu-TMs合金的形成能,发现加U 修正能够给出更接近于实验值的结果。
该研究表明正确模拟Cu-3d 和TM-d 带间的相互作用是准确表征Cu-TM合金的形成能等性质的关键,GGA+U 是一种高效调整TM-d 带至正确的能量分布范围的有效方法,在Cu-TMs体系中的修正为提升GGA在预测合金性质方面的应用提供了指导,如准确预测结构的形成能和有序-无序转变的临界温度等。
固体所博士后赵静为论文的第一作者,王贤龙研究员为通讯作者。该研究得到了国家自然科学基金、合肥物质院院长基金等项目的资助。